Organic Molecule generated from SMILES¶
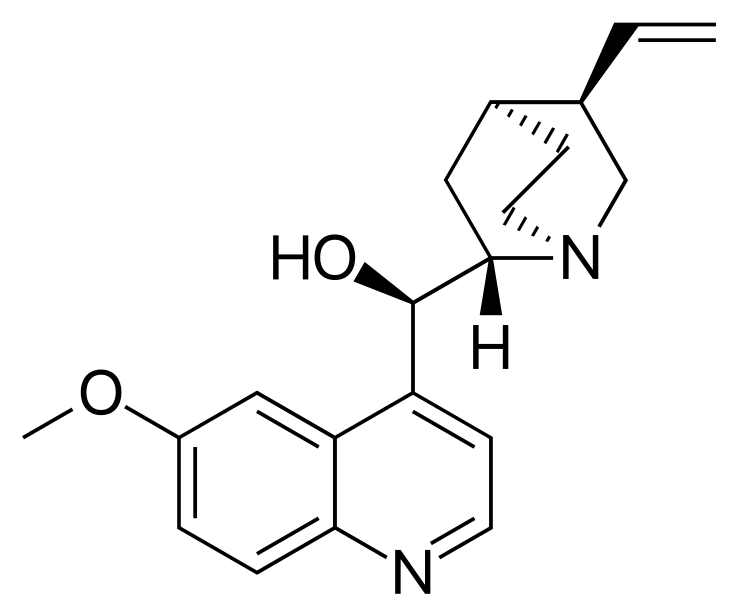
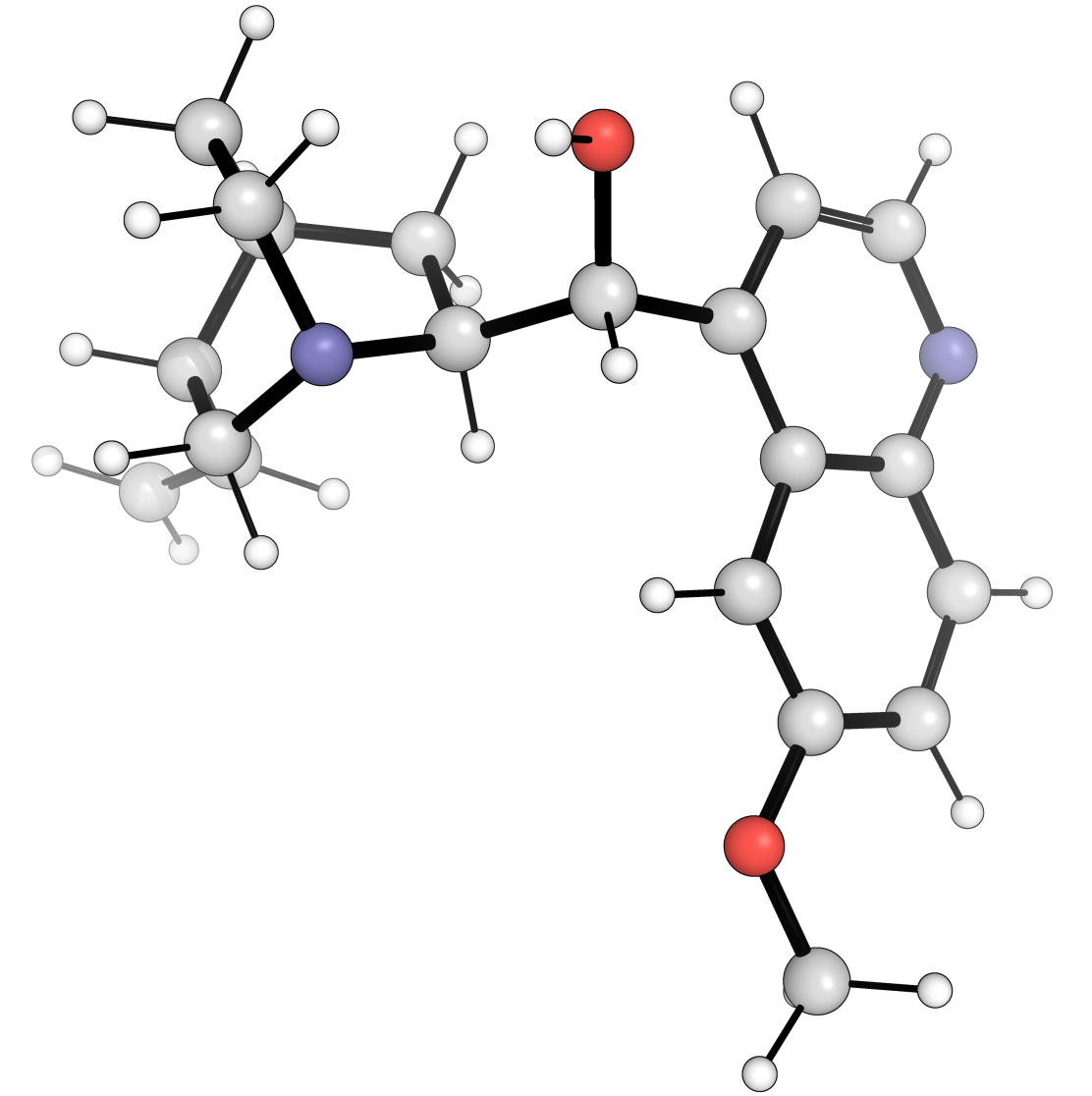
In the following examples we will be generating conformations of the quinine molecule.
SMILES |
|
COC1=CC2=C(C=CN=C2C=C1)[C@H]([C@@H]3C[C@@H]4CCN3C[C@@H]4C=C)O |
|
|
|
Conformer Generation¶
python -m aqme --csearch --smi "COC1=CC2=C(C=CN=C2C=C1)[C@H]([C@@H]3C[C@@H]4CCN3C[C@@H]4C=C)O" --destination quinine_folder --name quinine --program rdkit
Here we are specifying that we want to use rdkit for the conformer generation when we specify program='rdkit' and we are specifying the base name of the output files to be 'quinine'.
At this point we will have a new folder already created named 'quinine_folder' that contains a file named quinine_rdkit.sdf that contains all the conformers generated.
If we wanted to use fullmonte instead to generate the geometries then we just need to change the program parameter to 'fullmonte':
python -m aqme --csearch --smi "COC1=CC2=C(C=CN=C2C=C1)[C@H]([C@@H]3C[C@@H]4CCN3C[C@@H]4C=C)O" --destination quinine_folder --name quinine --program fullmonte
Minimizing the conformations¶
Back to our conformers generated using rdkit we might be interested in running an energy minization using XTB or ANI. To do so we will need the CMIN module.
python -m aqme --cmin --files "quinine_folder/*.sdf" --program ani --destination quinine_ani
Here 'destination' is the folder where the new optimized geometries will be generated, 'files' is a list of files that we want to minimize and 'program' is specifying that we want to run the minimizations using 'ani'.
Warning
Please notice that shell wildcard arguments need to be provided as strings.
--files "quinine_folder/*.sdf" should be provided instead of
--files quinine_folder/*.sdf. This feature might change in future to
follow the usual conventions.
Using csv files as input¶
Another way of providing the molecule to the program is by writing it into a csv file. Lets asume we have in our working directory the file 'ML_test.csv' with the following contents:
code_name,SMILES
quinine,COC1=CC2=C(C=CN=C2C=C1)[C@H]([C@@H]3C[C@@H]4CCN3C[C@@H]4C=C)O
With this file we can run the same conformer search that we run at the beggining with the following code:
python -m aqme --csearch --input ML_test.csv --program rdkit --destination quinine_folder
Using csv files allows specifying multiple molecules by their SMILES string in a single file and facilitates the reusability of the code. If we change the contents of 'ML_test.csv' to:
code_name,SMILES
methane,C
ethane,CC
propane,CCC
butane,CCCC
pentane,CCCCC
We can re-run the exact same code, and we will end with the conformers of each one of these molecules in the same directory. If we want to have them in a different folder we can simply change the destination:
python -m aqme --csearch --input ML_test.csv --program rdkit --destination alkanes_folder