QDESCP (descriptor generation)
Overview
The QDESCP module implemented in AQME enables the automated calculation of molecular and atom-centered descriptors from either SMILES strings or three-dimensional structures. In the current implementation, descriptor generation is performed through MORFEUS, which acts as a unified interface to obtain electronic and steric descriptors.
Electronic descriptors are extracted from xTB calculations, preferentially using the PTB method when available and GFN2-xTB for descriptors not supported at the PTB level. In parallel, steric descriptors are computed directly from the GFN2-xTB-optimized geometries.
The overall workflow is summarized in this figure:
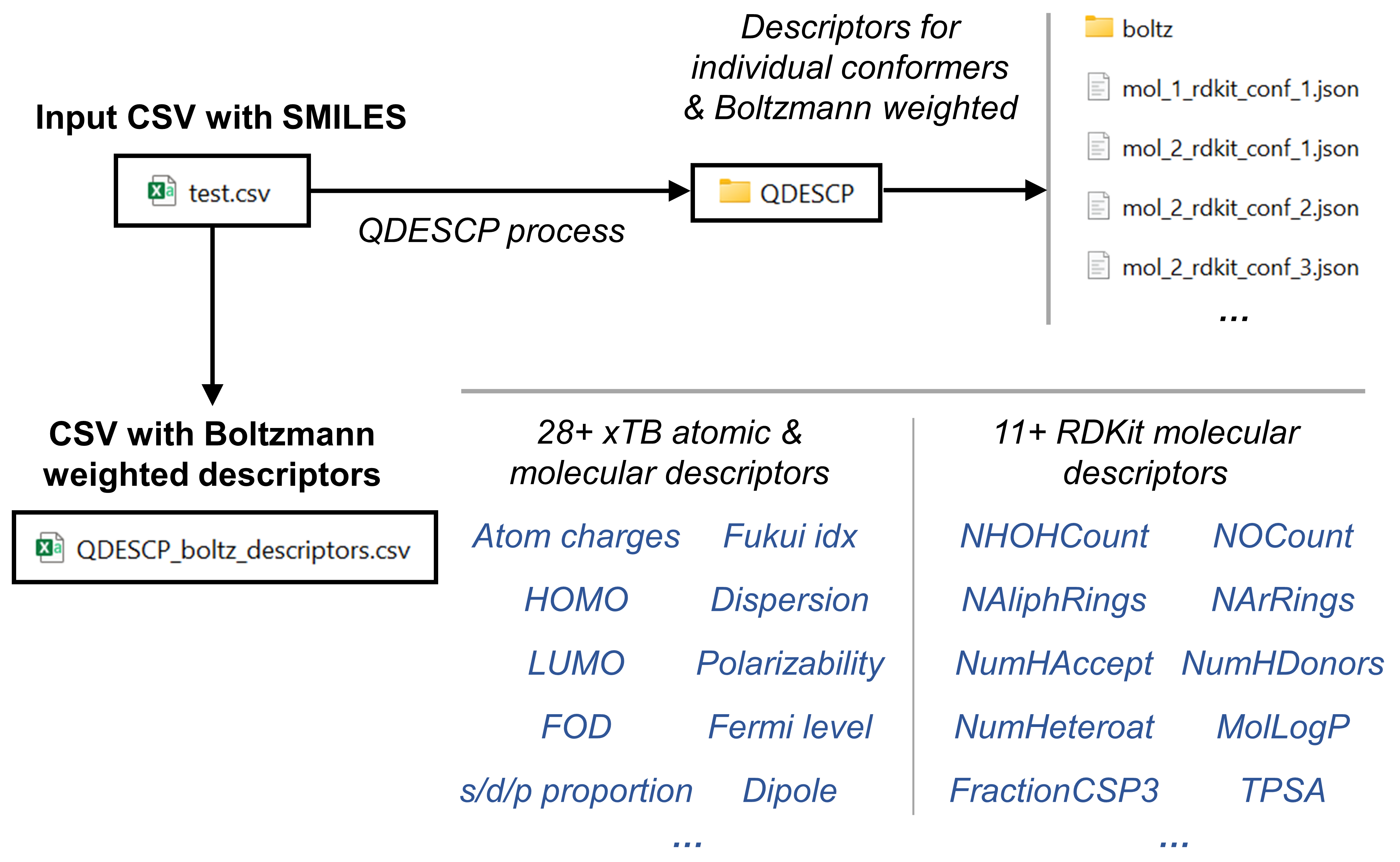
Workflow Description
1. Conformational search (if starting from SMILES)
When the starting point is a CSV file containing SMILES strings, conformational sampling is first performed using the CSEARCH workflow in AQME, typically employing RDKit (default) and optionally CREST.
The generated conformers are filtered using:
An energy window (ΔE ≤ 5 kcal mol⁻¹)
RMSD-based filtering
Butina clustering
This process retains a structurally diverse subset of low-energy conformers (up to five conformers per molecule), which are stored as SDF files.
If three-dimensional structures are provided directly (e.g., XYZ or SDF files), the conformational search step is skipped, and the structures are used directly for descriptor generation.
2. Geometry optimization with GFN2-xTB
The resulting conformers are subsequently optimized at the GFN2-xTB level. This option can be skipped using xtb_opt=False (or --xtb_opt False in command line).
3. Descriptor generation
Through MORFEUS, both molecular and atom-centered descriptors are obtained from the optimized geometries. Descriptors are first computed for each conformer and subsequently Boltzmann-weighted to obtain a single descriptor set per molecule.
Applicability
The workflow is applicable to both organic and organometallic systems, provided that the underlying xTB calculations converge reliably.
Warning
As with other semiempirical approaches, the quality of the resulting descriptors depends on the robustness of the electronic structure calculations. Systems involving:
Multireference character
Excited states (i.e., S₁, S₂, T₁...)
Aggregates or noncovalent complexes
should be interpreted with caution.
Output
The workflow produces CSV files containing Boltzmann-weighted descriptors corresponding to different descriptor subsets:
interpret (default)
denovo (reduced set)
full (extended set including RDKit features)
Descriptor Table
Note
All citations for the descriptors and formulae can be found in our publication describing the descriptor‑generation workflow.
The final descriptor set combines 21 molecular descriptors and 18 atomic descriptors, including:
xTB-based electronic descriptors (PTB method when available, if not GFN2-xTB)
MORFEUS steric descriptors
Type |
Full name |
Descriptor |
Definition / equation |
Units |
Method |
|---|---|---|---|---|---|
Molecular |
Highest occupied molecular orbital energy |
HOMO |
\(E_{\mathrm{HOMO}}\) |
eV |
PTB |
Molecular |
Lowest unoccupied molecular orbital energy |
LUMO |
\(E_{\mathrm{LUMO}}\) |
eV |
PTB |
Molecular |
HOMO–LUMO gap |
HOMO–LUMO gap |
\(E_{\text{gap}} = E_{\mathrm{LUMO}} - E_{\mathrm{HOMO}}\) |
eV |
PTB |
Molecular |
Ionization potential |
IP |
\(\mathrm{IP} = E(N-1) - E(N)\) |
eV |
GFN2 |
Molecular |
Electron affinity |
EA |
\(\mathrm{EA} = E(N) - E(N+1)\) |
eV |
GFN2 |
Molecular |
Molecular dipole moment magnitude |
Dipole module |
\(\text{-}\) |
debye |
PTB |
Molecular |
Solvent-accessible surface area |
SASA |
\(\mathrm{SASA} = \sum_i A_i\) |
Ų |
MORFEUS |
Molecular |
Dispersion surface area |
Dispersion area |
\(\text{-}\) |
Ų |
MORFEUS |
Molecular |
Dispersion volume |
Dispersion volume |
\(\text{-}\) |
ų |
MORFEUS |
Molecular |
Solvation free energy in water |
G solv. in H₂O |
\(\Delta G_{\text{solv}}\) |
kcal/mol |
GFN2 |
Molecular |
Hydrogen-bond contribution to solvation |
G of H-bonds H₂O |
\(\Delta G_{\text{HB}}\) |
kcal/mol |
GFN2 |
Molecular |
Fermi level |
Fermi-level |
\(E_F = \dfrac{E_{\mathrm{HOMO}} + E_{\mathrm{LUMO}}}{2}\) |
eV |
GFN2 |
Molecular |
Molecular polarizability |
Polarizability |
\(\text{-}\) |
a₀³ |
GFN2 |
Molecular |
Chemical hardness |
Hardness |
\(\eta = \mathrm{IP} - \mathrm{EA}\) |
eV |
GFN2 |
Molecular |
Chemical softness |
Softness |
\(S = \dfrac{1}{\eta}\) |
eV⁻¹ |
GFN2 |
Molecular |
Chemical potential |
Chem. potential |
\(\mu = -\dfrac{\mathrm{IP} + \mathrm{EA}}{2}\) |
eV |
GFN2 |
Molecular |
Electrophilicity index |
Electrophilicity |
\(\omega = \dfrac{(\mathrm{IP}+\mathrm{EA})^2}{8(\mathrm{IP}-\mathrm{EA})} = \dfrac{\mu^2}{2\eta}\) |
eV |
GFN2 |
Molecular |
Electrofugality |
Electrofugality |
\(\nu_{\text{electrofugality}} = \dfrac{(3\mathrm{IP}-\mathrm{EA})^2}{8(\mathrm{IP}-\mathrm{EA})} = \mathrm{IP} + \omega\) |
eV |
GFN2 |
Molecular |
Nucleofugality |
Nucleofugality |
\(\nu_{\text{nucleofugality}} = \dfrac{(\mathrm{IP}-3\mathrm{EA})^2}{8(\mathrm{IP}-\mathrm{EA})} = -\mathrm{EA} + \omega\) |
eV |
GFN2 |
Molecular |
Fractional occupation density |
Total FOD |
\(\text{-}\) |
e |
GFN2 |
Molecular |
Singlet–triplet energy gap |
S₀–T₁ gap |
\(\Delta E_{S0\text{-}T1} = E_{T1} - E_{S0}\) |
kcal/mol |
GFN2 |
Atomic |
Atomic hydrogen-bond contribution to solvation |
H-bond H₂O |
\(\Delta G_{\text{HB},i}\) |
kcal/mol |
GFN2 |
Atomic |
Mulliken partial charge |
Partial charge |
\(q_i\) |
e |
PTB |
Atomic |
Atomic dipole moment magnitude |
Dipole moment |
\(\text{-}\) |
debye |
PTB |
Atomic |
Atom solvent accessible surface area |
Atom SASA |
\(A_i\) |
Ų |
MORFEUS |
Atomic |
Atomic dispersion descriptor |
Atom dispersion |
\(\text{-}\) |
Ų |
MORFEUS |
Atomic |
Percent buried volume |
Buried volume |
\(\text{-}\) |
% |
MORFEUS |
Atomic |
Pyramidalization parameter |
Pyramidalization |
\(P = \sin(\theta)·\cos(\alpha)\) |
\(\text{-}\) |
MORFEUS |
Atomic |
Pyramidalization angle |
Pyramidaliz. volume |
\(P = \sqrt{360^\circ - \sum_i \theta_i}\) |
° |
MORFEUS |
Atomic |
Fukui nucleophilic index |
Fukui+ |
\(f^+ = q_N - q_{N+1}\) |
\(\text{-}\) |
GFN2 |
Atomic |
Fukui electrophilic index |
Fukui− |
\(f^- = q_{N-1} - q_N\) |
\(\text{-}\) |
GFN2 |
Atomic |
Radical Fukui index |
Fukui_rad |
\(f_{\mathrm{rad}} = \dfrac{q_{N-1} - q_{N+1}}{2}\) |
\(\text{-}\) |
GFN2 |
Atomic |
Dual Fukui descriptor |
Fukui dual |
\(f^{(2)} = f^+ - f^-\) |
\(\text{-}\) |
GFN2 |
Atomic |
Local electrophilicity |
Electrophil. |
\(l_\omega = -\dfrac{\mu}{\eta} f + \tfrac{1}{2}\dfrac{\mu}{\eta^2} f^{(2)}\) |
\(\text{-}\) |
GFN2 |
Atomic |
Normalized electrophilicity |
Normaliz. electrophil. |
\(\omega_i = \omega\ · f_i^+\) |
eV |
GFN2 |
Atomic |
Normalized nucleophilicity |
Normaliz. nucleophil. |
\(N_i = -\mathrm{IP}\ · f_i^-\) |
eV |
GFN2 |
Atomic |
Atomic polarizability |
Atom Polarizability |
\(\text{-}\) |
a₀³ |
GFN2 |
Atomic |
Atomic FOD population |
Atom FOD |
\(\text{-}\) |
e |
GFN2 |
Atomic |
Coordination number |
Coord. numbers |
\(\text{-}\) |
\(\text{-}\) |
GFN2 |