Diels-Alder reactions
Along the steps of this example workflow we will show how to:
Generate different conformers of molecules and noncovalent complexes using CREST
Generate the inputs for Gaussian geometry optimizations and frequency calcs (B3LYP/def2TZVP)
Fixing errors and imaginary frequencies of the output LOG files
Generate ORCA inputs for single-point energy corrections (SPC) using DLPNO-CCSD(T)/def2TZVPP
Calculate the Boltzmann weighted thermochemistry using with GoodVibes at 298.15 K
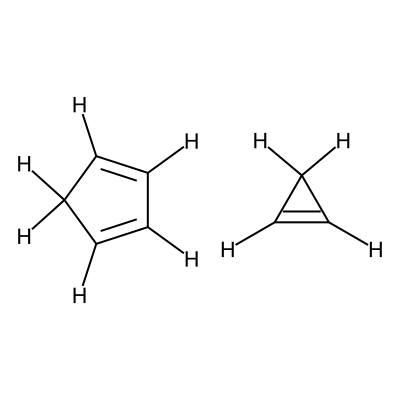
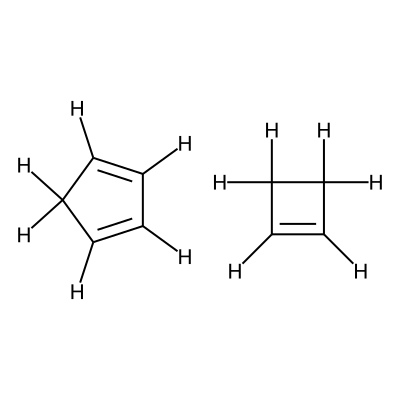
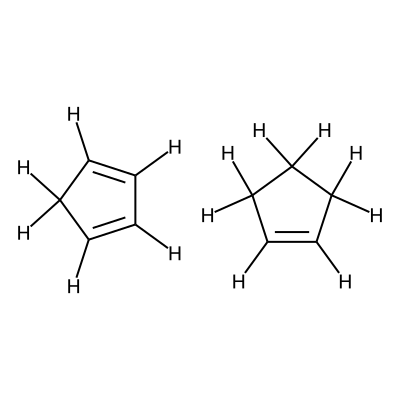
Specifially in this workflow we will calculate the free energy profile for the Diels-Alder reaction for three pairs of reactants shown below:
Reactants 1 |
Reactants 2 |
Reactants 3 |
C1=CC=CC1.C1=CC1 |
C1=CC=CC1.C1=CCC1 |
C1=CC=CC1.C1=CCCC1 |
|
|
|
Note
A jupyter notebook containing all the steps shown in this example can be found in the AQME repository in Github or in Figshare
Step 1: Determining distance and angle constraints for TSs
In the following examples we need the mapped SMILES to set up the constraints. We can either use rdkit for it in python, in a jupyter notebook (see python example ) or we can write the SMILES by ourselves (not recommended).
We visualize the first pair of reactants to be able to set up the constraints.
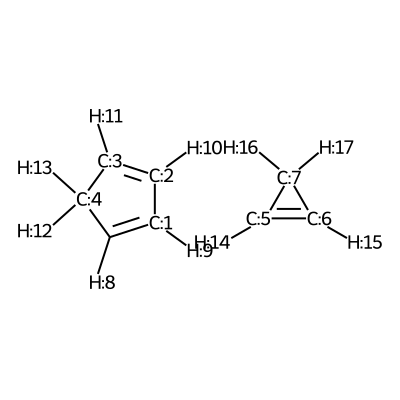
C1([H:8])=[C:1]([H:9])[C:2]([H:10])=[C:3]([H:11])[C:4]1([H:12])[H:13].[C:5]1([H:14])=[C:6]([H:15])[C:7]1([H:16])[H:17]
According to the image we will add the following constraints to the CSV, in the
constraints_dist column we will include [[3,5,2.35],[0,6,2.35]]
We visualize the second pair of reactants to be able to set up the constraints.
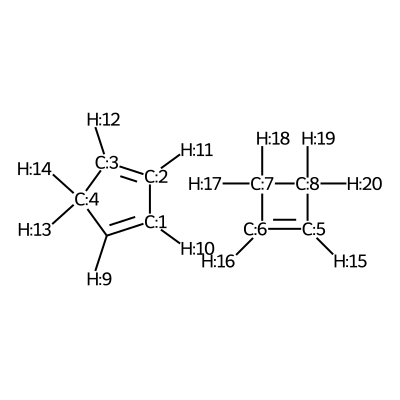
C1([H:9])=[C:1]([H:10])[C:2]([H:11])=[C:3]([H:12])[C:4]1([H:13])[H:14].[C:5]1([H:15])=[C:6]([H:16])[C:7]([H:17])([H:18])[C:8]1([H:19])[H:20]
According to the image we will add the following constraints to the CSV, in the
constraints_dist column we will include [[3,6,2.35],[0,5,2.35]]
Warning
Although the atoms 5 and 6 are equivalent, we have observed that if we use
the same ordering as in the previous reaction for the constraints the TS
won't be found (i.e. with [[3,5,2.35],[0,6,2.35]]) whereas when we
use the constraints as shown in the example the TS is found.
We visualize the third pair of reactants to be able to set up the constraints.
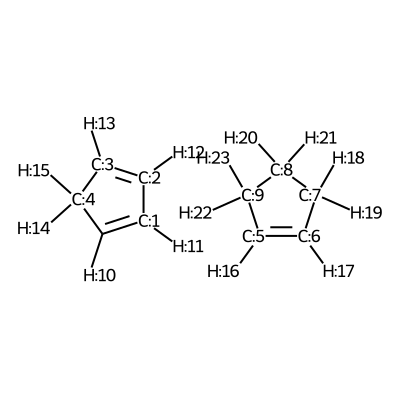
C1([H:10])=[C:1]([H:11])[C:2]([H:12])=[C:3]([H:13])[C:4]1([H:14])[H:15].[C:5]1([H:16])=[C:6]([H:17])[C:7]([H:18])([H:19])[C:8]([H:20])([H:21])[C:9]1([H:22])[H:23]
According to the image we will add the following constraints to the CSV, in the
constraints_dist column we will include [[3,5,2.35],[0,6,2.35]]
Step 2: CSEARCH conformational sampling
With the previous step we can now create a csv file containing all the molecules and noncovalent complexes to calculate, which will have the following contents:
SMILES,code_name,constraints_dist
C1=CC=CC1,Diene,
C1=CC1,Do1,
C1=CCC1,Do2,
C1=CCCC1,Do3,
C1([H:8])=[C:1]([H:9])[C:2]([H:10])=[C:3]([H:11])[C:4]1([H:12])[H:13].[C:5]1([H:14])=[C:6]([H:15])[C:7]1([H:16])[H:17],TS1,"[[3,5,2.35],[0,6,2.35]]"
C1([H:9])=[C:1]([H:10])[C:2]([H:11])=[C:3]([H:12])[C:4]1([H:13])[H:14].[C:5]1([H:15])=[C:6]([H:16])[C:7]([H:17])([H:18])[C:8]1([H:19])[H:20],TS2,"[[3,6,2.35],[0,5,2.35]]"
C1([H:10])=[C:1]([H:11])[C:2]([H:12])=[C:3]([H:13])[C:4]1([H:14])[H:15].[C:5]1([H:16])=[C:6]([H:17])[C:7]([H:18])([H:19])[C:8]([H:20])([H:21])[C:9]1([H:22])[H:23],TS3,"[[3,5,2.35],[0,6,2.35]]"
[C@H]1(C2C=CC3C2)[C@@H]3C1,P1,
[C@H]12[C@@H](C3C=CC2C3)CC1,P2,
[C@H]1(C2C=CC3C2)[C@@H]3CCC1,P3,
Now we can proceed to the conformer generation:
python -m aqme --csearch --input example2.csv --program crest --cregen --cregen_keywords "--ethr 0.1 --rthr 0.2 --bthr 0.3 --ewin 1" --nprocs 12
Step 3: Creating Gaussian input files for optimization and frequency with QPREP
We first create the input files of the transition states
python -m aqme --qprep --program gaussian --mem 32GB --nprocs 16 --files "CSEARCH/TS*crest.sdf" --qm_input "B3LYP/def2tzvp opt=(ts,calcfc,noeigen,maxstep=5) freq=noraman"
Now we create the input files of the minima (intermediates, reagents and products)
python -m aqme --qprep --program gaussian --mem 32GB --nprocs 16 --files "CSEARCH/D*.sdf" --qm_input "B3LYP/def2tzvp opt freq=noraman"
python -m aqme --qprep --program gaussian --mem 32GB --nprocs 16 --files "CSEARCH/P*.sdf" --qm_input "B3LYP/def2tzvp opt freq=noraman"
Step 4: Running Gaussian inputs for optimization and frequency calcs externally
Now that we have generated our gaussian input files (in the QCALC location of Step 3) we need to run the gaussian calculations. If you do not know how to run the Gaussian calculations in your HPC please refer to your HPC manager.
As an example, for a single calculation in Gaussian 16 through the terminal we would run the following command on a Linux-based system:
g16 myfile.com
Step 5: QCORR analysis
python -m aqme --qcorr --files "QCALC/*.log" --freq_conv "opt=(calcfc,maxstep=5)" --mem 32GB --nprocs 16
Step 6: Resubmission of unsuccessful calculations (if any) with suggestions from AQME
Now we need to run the generated COM files (in fixed_QM_inputs) with Gaussian like we did in Step 4
After the calculations finish we check again the files using QCORR
python -m aqme --qcorr --files "QCALC/failed/run_1/fixed_QM_inputs/*.log" --isom_type com --isom_inputs "QCALC/failed/run_1/fixed_QM_inputs" --nprocs 16 --mem 32GB
Step 7: Creating DLPNO input files for ORCA single-point energy calculations
python -m aqme --qprep --program orca --mem 16GB --nprocs 8 --files "QCALC/success/*.log" --suffix DLPNO --destination SP --qm_input "DLPNO-CCSD(T) def2-tzvpp def2-tzvpp/C
%scf maxiter 500
end
% mdci
Density None
end
% elprop
Dipole False
end"
Step 8: Running ORCA inputs for single point energy calcs externally
Now we need to run the generated inp files (in sp_path) with ORCA (similarly to how we did in Step 4)
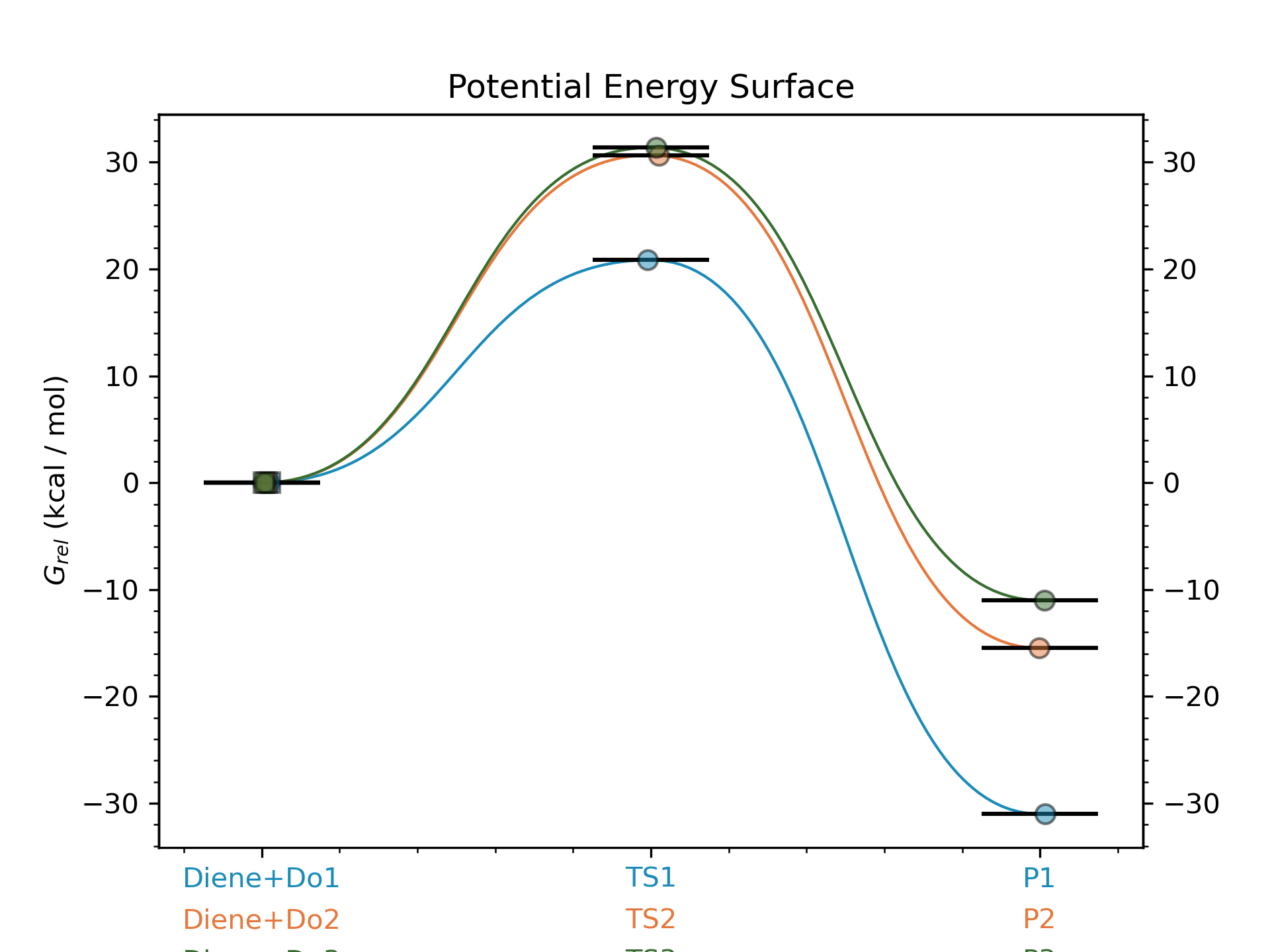
Step 9: Calculating PES with goodvibes
for this step we will need to have a yaml file to use as input for goodvibes. The contents of the yaml file are:
--- # PES
# Double S addition
Reaction1: [Diene+Do1, TS1, P1]
Reaction2: [Diene+Do2, TS2, P2]
Reaction3: [Diene+Do3, TS3, P3]
--- # SPECIES
Diene : Diene*
Do1 : Do1*
TS1 : TS1*
P1 : P1*
Do2 : Do2*
TS2 : TS2*
P2 : P2*
Do3 : Do3*
TS3 : TS3*
P3 : P3*
--- # FORMAT
dec : 1
units: kcal/mol
dpi : 300
color : #1b8bb9,#e5783d,#386e30
With this file we can now generate the profile.
mkdir -p GoodVibes_analysis
cp SP/*.out GoodVibes_analysis/
cp QCALC/success/*.log GoodVibes_analysis/
cd GoodVibes_analysis
python -m goodvibes --xyz --pes ../pes.yaml --graph ../pes.yaml -c 1 --spc DLPNO *.log
cd ..